It was quite a shock to perform "control" experiments wherein supposedly "wild type" E. coli was streaked out on agar plates containing ampicillin, only to discover that quite a few colonies formed, which meant of course some of the bacterial cells had acquired the gene for antibiotic resistance. When E. coli that has never been exposed to ampicillin is used in the control, it is invariably true that no colonies ever form. So, our "wild type" cells had obviously been exposed to ampicillin at some point, and found a way to start making an ampicillin-destroying enzyme. The explanation lies in the techniques used in the lab: plastic pipettes are routinely rinsed in water and then sterilized with alcohol before being allowed to dry in their original paper sleeve. (Note: we are reducing our plastics waste while saving money on new pipettes!) There appears to be no down side to this technique in terms of sterility, since poured agar plates, prepared utilizing alcohol-sterilized plastic pipettes, show no signs of extraneous growth after several weeks held at room temperature. But, after segregating amp and non-amp plastic labware, the problem of amp-resistance creeping in to wild type E. coli disappeared. This means that even rinsed pipettes carried over enough ampicillin to give a few cells the ability to become amp-resistant, a tiny amount of antibiotic by any standard. It also gives rise to the question: how on earth can E. coli "learn" to become amp-resistant in a such short time (a week or two)? Do the cells constantly adapt to foreign molecules and find ways to metabolize them? Does this have implications regarding a cell's "molecular immune system" to say nothing about antibiotic resistance in general?
It would benefit the planet greatly to make a "wonder organism" that can:
a) grow in arid conditions or in seawater
b) produce large amounts of energy-rich material (such as oil)
c) be harvested easily and processed cheaply to recover said energy-rich material.
The above sounds fantastic: all we have to do is get busy on the genetic engineering part to transform, say, algae, into this wonder organism. But, there seems to be a catch. Bacteria that grow in the presence of an antibiotic only survive because they always produce an enzyme that destroys the antibiotic. Take away the antibiotic, and they soon stop making the enzyme. Future generations are susceptible to the antibiotic--in effect, the bacteria have to "relearn" to destroy it (see above). Or, they can be transformed with fresh plasmid which harbors the gene for antibiotic destruction, and they start to survive. If a separate useful protein is coded by the DNA that codes for antibiotic destruction, then in the process of maintaining antibiotic resistance, the bacteria will also make the useful protein if the gene for making it is turned on. This appears to be true for 50-100 generations. It has been found that if a bacterium which has been engineered to make GFP is maintained by repeatedly transferring it to fresh culture medium (that is, keeping it alive for well more than 100 generations), eventually it stops producing GFP, even though it is still making the protein which destroys antibiotic. This is interpreted as the bacterium "learning" to stop wasting its metabolic energy on making GFP since the GFP confers no advantage to the bacterium--unlike the antibiotic destroying enzyme, which of course is crucial for survival if antibiotic is present. In short, without environmental pressure, wasteful genes are eventually shut down, evidently as part of an overall survival mechanism, which strives for maximum efficiency. This of course has implications for the photosynthetic wonder organism we started with, since clearly making oil does not confer a benefit to the organism--and tends to put a damper on "getting busy" doing the genetic engineering, if all that work is going to be ignored by the organism, eventually.
Phenotype refers to the outward, visible traits an organism displays which are derived from inheritance. Thus, the size of an elephant's ears would be a phenotype, but an elephant's ability to stand on its front legs after it has been trained would not be a phenotype. (It could be argued that an elephant's ability to be trained at all is a phenotype, distinguishing it from other elephants and other species.) The part that gets inherited is of course carried by genes, and so the specific genetic attributes that an organism has (mostly DNA sequences) are called genotypes. Generally speaking, a change in the genotype can result in a new phenotype, if the genetic changes manifest themselves in observable ways. Often, changes in genotype are "silent" and result in no apparent new phenotype. But if a new phenotype is observed, it is a good bet a corresponding genotype change is present. This is why picking E. coli colonies that look different from the colonies that have not undergone a genetic change is so fruitful, and usually results in finding a new genotype. Note that in these cases, the changes are in the genes carried by plasmids, making it a relatively simple matter to isolate the genes and characterize them (that is, obtain their DNA sequences).
The protocol which has been adopted in this laboratory was born partly out of a desire to use the old-school DNA isolation method, which allows one to actually see a DNA precipitate (and costs practically nothing), and partly to increase the chances that DNA sent off to be sequenced actually has the expected sequence. In addition, growing E. coli in overnight cultures to obtain enough cells for DNA isolation is simple enough that the two step procedure boils down to adding some extra time to the process of getting results, with the advantage of much greater confidence in the results, without wasting too much money sequencing useless DNA. The protocol involves first transforming E. coli with plasmids that have been constructed for the purpose of a particular experiment, streaking out the transformed cells, and selecting colonies which appear "different." Different generally means a different level of brightness observed in the green fluorescence, sometimes brighter and sometimes less bright, compared to the unchanged organism. The plasmid construction protocols, which vary, usually involve placing new DNA into the plasmid and also the possibility of reinserting the old DNA from the original plasmid back into its original place, which gives back the same genotype (and phenotype) as was started with, i.e. the transformed but unchanged organism. Now, once a colony is picked, it is suspended in 2 ml of culture medium and allowed to grow overnight. At this point, that DNA could be isolated using a kit and sent off for sequencing, with a real chance the unchanged organism had been selected, since often, the phenotype changes are quite subtle and hard to distinguish. However, the DNA is isolated via a non-kit protocol (see "Molecular Cloning," Sambrook, J. and Russell, D.W. 3rd ed., Cold Spring Harbor Press, 2001) which works every time to give DNA which transforms cells easily. This DNA is not pure enough for sequencing or for further plasmid manipulation. In the second step, the non-kit DNA is used to transform cells, these are streaked out, and the phenotype is often confirmed--namely, a "bright" clone will appear bright, or a "dim" one will appear dim, provided the original "different" colony was picked and accurately characterized in the first place. Colony selection occurs once again (see the side bar), only this time the colony is grown in 4 ml of culture overnight, and the DNA is kit-isolated, giving material pure enough for sequencing or for further plasmid manipulations.
Sidebar: One of the advantages of the two-step protocol is that strange DNA behavior can be discovered and filtered out. For example, the plasmid construction method sometimes results in a triple insert, which usually shuts down GFP production, but sometimes gives fluorescent colonies (the mechanism is unclear, but GFP expression appears to sometimes be "on" and sometimes be "off.") At any event, if a dim colony is picked and the 2nd step gives colonies that are not all the same, with some obviously brighter than others, then this is taken as evidence of a triple insert. Note that triple inserts from this lab have been sequenced, supporting this contention.
After using E. coli in the lab for several months, it became evident that it can take on characteristics that are peculiar to the lab environment. Its ability to acquire ampicillin resistance after growing in the presence of trace amounts of the antibiotic has been noted above (see "Resistance"). When these same cells are streaked out on fresh agar (without amp) and then several colonies are picked and again streaked out on agar, the resultant cells take on new characteristics: a) they become susceptible to amp, and b) certain cultures take on the beneficial property of adhering less tightly to the plastic centrifuge tubes used in their manipulation. This is useful, since it is often necessary to re-suspend cells in media or some other solution, and it is quite tedious to vortex tubes (especially by hand) and have the cells remain stuck to the plastic in the pellet at the bottom of the tube for extended periods. It also may be important to suspend cells "gently" once they become competent, since their cell walls are now fragile. The new "non-sticky" phenotype, however, re-suspends easily, greatly speeding up most cell manipulations. So, if you find your E. coli becoming sticky, it's worth streaking them out (and maybe even letting them acquire amp resistance, and then losing it over a few generations first), and see if a colony can be isolated which is less sticky.
-Note
added after using "non-sticky" bacteria for several
months:
It seems likely that yet another peculiarity of our lab has given us the non-sticky phenotype. Namely, our liquid cultures are not shaken. This means the E. coli have no reason to try to affix themselves to the sides of the flask, and instead move themselves via their flagella to attain the environment they want (they are "free range" if you like). Indeed, overnight cultures have only a small amount of cells settled on the bottom of the flask, and the cultures are very turbid. We have good growth with high yields of DNA and protein. In the gut, there are mechanisms for forming biofilms which prevent cells from simply washing away--so possibly E. coli can also change its cell surface in such a way as to make itself "sticky" if it finds itself moving around too much--and our cells have turned this off.
It seems likely that yet another peculiarity of our lab has given us the non-sticky phenotype. Namely, our liquid cultures are not shaken. This means the E. coli have no reason to try to affix themselves to the sides of the flask, and instead move themselves via their flagella to attain the environment they want (they are "free range" if you like). Indeed, overnight cultures have only a small amount of cells settled on the bottom of the flask, and the cultures are very turbid. We have good growth with high yields of DNA and protein. In the gut, there are mechanisms for forming biofilms which prevent cells from simply washing away--so possibly E. coli can also change its cell surface in such a way as to make itself "sticky" if it finds itself moving around too much--and our cells have turned this off.
If a plasmid has two identical restriction sites close together, and moreover, if these sites are on either side of an important piece of DNA, then a single restriction enzyme can be used to cut out the DNA, and then see what the the new DNA (absent the important bit) will code for. This was done for a plasmid coding for GFP, where the final stop signal (TAA) was between two SacI sites (in the boxes at right). In the early days, we were more interested in what could be done at all, rather than what could be done that was important. So as part of a training exercise, E. coli harboring the plasmid was grown in sufficient quantity, the plasmid DNA was isolated with a mini-prep kit, the DNA was cut with SacI, and then ligated with T4 ligase. Finally, it was used to transform a fresh batch of competent E. coli, and the cells were observed on agar plates. We were very pleased to see two phenotypes. One looked like the E. coli we started out with, which was making normal GFP. The other colonies appeared "dim" with attenuated green fluorescence. It was relatively easy to tell the two apart, but the two step procedure identified above came in handy: the DNA from the "dim" colonies was isolated and used to transform cells, and when these were streaked out, the brightness of 10-50 colonies was easier to compare to plates of 10-50 colonies making normal GFP, than trying to compare the brightness of individual colonies all on one plate. Finally, the new plasmid was isolated using a kit and sent off for sequencing, which showed the DNA had only one SacI site and the TAA was gone, as illustrated at right. (The next stop signal happened to be about 60 nucleotides downstream, which gave a GFP with a long tail with no apparent biological function). The really great thing about molecular biology is that every experiment can turn out to be useful. So even though the SacI cutting experiment described here was done initially to make sure various protocols were working, the resulting plasmid and the phenotype created in E. coli became a workhorse in the lab. If the now unique SacI site is used along with another site downstream of it, DNA inserts with their own stop signals can be installed which generally give bright phenotypes, and are usually easy to distinguish from unchanged colonies during routine insert experiments.
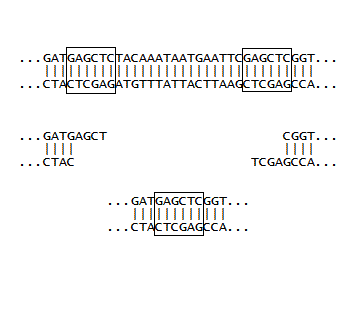
A few initial inserts were designed with the goal of maximizing the number of coding nucleotides, and minimizing the number of bases used for joining the insert DNA to the plasmid DNA. This was done by building in restriction site overhangs directly into the inserts so that the overhangs would complement the overhangs left after the plasmid was treated with appropriate restriction enzymes (namely, SacI for the DNA fragment at bottom right). In order for this to work, a 5'-phosphate needs to be installed on the insert, since the synthetic oligos are missing this phosphate, and it is required during the ligation step. The enzyme required for this installation is called polynucleotide kinase, which was duly ordered and used. However, in our hands this reaction failed. Thus it became more expedient to install complete restriction sites at both ends of the inserts (one is shown as a SacI site in the top picture). This requires that they are cut with the same enzymes used to cut the plasmids. This leaves 5'-phosphates where you need them, making ligation convenient. Note the two CC's at the beginning are enough "buffer" to make it possible for the restriction enzyme to cut, and the greater strength of the CG interaction (as opposed to an AT interaction) help in the formation of double stranded DNA. From the picture one can see that two additional coding nucleotides are made available with a built in overhang in the bottom sequence (because the CC is not needed). However, the complete restriction site method was used for inserts, since performing the restriction enzyme digestion alongside the plasmid digestion was very convenient, and this method proved to be very reliable. Additionally, a novel assembly method (detailed in later pages) greatly increases the effective lengths of inserts, making the conservation of coding nucleotides less essential.
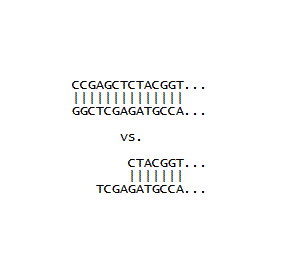
Quote from a chemist overheard at a research conference:
"It is a severe indictment of the
human intellect that we
have to rely on combinatorial techniques to design molecules."
have to rely on combinatorial techniques to design molecules."
This can be extended to protein and peptide design, in that often a random mutation is what gives the most useful or interesting result (and not one arrived at by design), and indeed, the use of artificial intelligence to "design" proteins is in the same league as relying on random chance, since it is by no means clear that the machine in "machine learning" is willing or capable to "teach" us how it arrived at a given result. Still, this pessimism aside, there is room for hope that we can partner with computers to tackle the problem of protein design, and even the allied issue of protein folding. This is because computers make it possible to visualize proteins and things like binding pockets. Looking at a ribbon structure of a protein can be compared to looking at a cloud from the ground, and this can be compared to looking at individual atoms of a part of the protein to viewing a cloud from a small airplane flying into it or around it: the details are clearly lost when viewing "from a distance." It is with this spirit that protein crystal structures can be downloaded and then viewed, as a whole or in part, with as much detail as necessary, to arrive at an understanding of the structure, as well as to develop perhaps some small ability to place individual amino acids in a peptide in order to design it to accomplish some particular goal. The image below is an approximate rendering of strands 10 and 11 of GFP, along with the "10-11 loop" which connects them. The loop in fact is empty in the picture, but is filled with an R group in the actual protein. One can dig deeper and find many hydrogen bonds and salt bridges that help stabilize the loop as well as the alignment of the strands. Thus, it should be possible to learn some design principles by imitating nature in specific instances, such as in the picture below. This can be pursued with the appreciation that it may not lead to a complete understanding of protein structure (or folding), but could at least lead to the design of useful and functional peptides.
